
Numerous studies have drawn attention to the circulation of WNV in Africa and Europe, but little is known about the genetic relationships and the introductory event dynamics of WNV L1 and L2 European and African strains10,11,18,19. In this study, we used phylogenetic and phylogeographic inference to uncover the origins, genetic relationships, dispersal history, and geographic patterns of most of the WNV L1 and L2 strains that have circulated and are circulating in both continents.
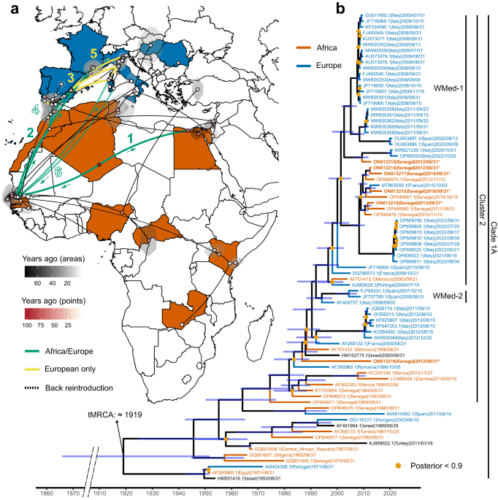
Of the two, WNV L1 is by far the lineage that appears to have the more complex history, with multiple clades, clusters and sub-clusters, and genetic flows among countries and continents around the world. Previous studies have described three major WNV L1 clades: 1A, which included strains from Africa, Europe, the Middle East, Asia, and America; 1B, which contained Kunjin virus, a virus circulating in Australia; and 1C, which comprised strains from India13,14,22. Within Clade 1A, further subclassifications of WNV L1 have been made in the past13,23,24,25, with seven clusters (clusters 1 to 7) recognised, providing evidence of the very complex structure of this lineage. Our phylogenetic analysis is consistent with the previous classification and helps to provide new insights into the hierarchical structure of WNV L1 Clade 1A (Fig. 1 and Supplementary Fig. 1).
According to our analyses, the WNV strains included in most of the Clade 1A clusters (2, 3, 4, 6, and 7) are likely to originate from Africa, more specifically from West Africa. Clade 1A clusters 2, 3, 4, 6, and 7 are in fact rooted by the two ancient sequences from Nigeria and Senegal included in cluster 5 (Fig. 1 and Supplementary Fig. 1). More interestingly, cluster 2, which includes most of the European strains, appears to be rooted by the Senegal strain of 1989 (Genbank OP846971) (Fig. 1). This means that the first European introduction probably originated in Senegal around the 1990s, as previously reported by Ndione et al.15. This hypothesis is further supported by our phylogeographic inference which not only confirms the middle of North and West Africa as the location of the common ancestor of all WNV L1 Clade 1A strains but also traces its origin back to the 19th/20th century. From this ancestral site, the virus probably spread towards Egypt and Senegal, the latter being at the origin of all WNV strains within the WMed sub-cluster (20th – 21st centuries) (Fig. 1).
The 1996 Morocco strain (Genbank AY701412) at the root of the WMed 1 and 2 subtypes implies a Moroccan origin of most of the Western-Mediterranean European WNV L1 strains. This appears to have occurred around the 1990s and reinforces the previously hypothesised assumption of the existence of a genetic flow between North of Africa (Morocco) and Italy and France [Origin and evolution of West Nile virus lineage 1 in Italy, unpublished manuscript]26.
These findings confirm the presence of a corridor between Senegal, Morocco, and Western-Mediterranean European countries, such as Portugal, Spain, France, and Italy (Fig. 1). According to our phylogenetic inference, this is not a one-way corridor as incursions of WNV strains from Europe into Africa have also been shown to occur. The WMed 1 and WMed 2 groups within the WMed sub-cluster which includes European, Moroccan (2003) and Senegalese (2012 − 2018) strains, are sister of the 2000 French strain (Fig. 1 and Supplementary Fig. 1). Our analyses indicate that the 2003 Morocco strain originated from an area located in the South of Spain, from which sequences from Portugal (2004) and France (2004) also derived. This is the first time that such an evolutionary relationship has been disclosed. The genetic similarities observed between i) the 2012-2018 Senegalese and 2015 French strains15, all of which are closely related to the group of 2021-22 Italian sequences, and ii) the 2020-2022 Italian and 2012 Senegalese strains and those from Italy (2008 and 2011) and Spain (2020) (Fig. 1), further support this new reconstructive theory.
The evolutionary history of WNV L2 strains is much simpler to describe, with few introductory events from South Africa to Europe and no genetic flow in the opposite direction. Our maximum-likelihood phylogeny and molecular clock analysis stress the presence of several African WNV L2 clades with common origins (Fig. 2 and Supplementary Fig. 2). Historically, WNV L2 has circulated in Sub-Saharan Africa and Madagascar27, before spreading to Europe14,28. Malagasy strains appear far apart from all the other strains included in this analysis, highlighting the presence of a local cycle probably sustained by resident birds and vector-competent mosquitoes on the island, and independent of the annual movements of migratory birds29. The evolution of permanent and stable local cycles, established after rare incursions into new areas, appears to be the main characteristic of the WNV L2 strains. Often, the genetic similarity of WNV L2 clades and clusters appears to be related to the specific geographical areas of circulation. This is supported by the fact that most of our WNV L2 strains cluster within the same geographical areas, as shown by i) Clade 2a and c, containing only strains from Madagascar; ii) Clade 2b, with most strains from South Africa and Namibia; and iii) Clade 2d, including cluster 1, composed of Southern and Central African strains; clusters 3 and 4, composed only of Senegal and Southern African (South Africa, Namibia, Uganda) strains, respectively; and cluster 5, with most Central-Southern European strains grouped together (Fig. 2 and Supplementary Fig. 2).
Interestingly, our phylogenetic inference shows that Clades 2c and 2d are a sister group of Clade 2b, which includes the first strain obtained in South Africa in 1958 (Supplementary Fig. 2). Furthermore, Clade 2d, which includes many African and European strains, appears to be rooted by the 1958 DRC strain, with high posterior support (Fig. 2). In particular, cluster 5 within Clade 2d including the Central-Southern European strains, all rooted by the Hungarian strain of 2004, are closely related to the South African cluster 4. These results stress the existence of a possible connection between South Africa and Europe. Our phylogeographic inference also supports this hypothesis, showing that the common ancestor of WNV L2 is likely to have originated in South Africa (Fig. 2), as already suggested in30. From there, WNV L2 probably spread among African countries, reaching Madagascar, DRC, CAR, Uganda, and Senegal in the 20th century, and to Europe in the 21st century. The virus, probably carried by long-distance migratory birds, was introduced into Hungary in 2004, where it was first detected, and then spread to many European countries (see Fig. 2), as previous studies have shown14,31.
Overall, while the evolutionary history of WNV L1 is characterised by a complex behaviour possibly due to the constant connections between different continents, WNV L2 shows only one main independent introduction from Southern African countries (cluster 4) to Europe (cluster 5). Once in Europe, the lineage began to spread all over the continent, founding favourable eco-climatic conditions, which allowed it to become endemic and a serious public health concern in numerous regions and countries28,32. Our phylogenetic and phylogeographic reconstructions also suggest a second introduction of minor importance from Southern African countries to Europe, supported by the existence of a separate cluster 2 within Clade 2d, which includes three European strains (Italy/2014, Romania/2014, Hungary/2017) closely related to the strains of CAR (1993) and South Africa (1958 and 1977) of cluster 1.
According to these findings, WNV L1 and L2 strains have had very different eco-epidemiological and genetic evolutionary features. The more complex genomic heterogeneity of WNV L1 strains may be a consequence of the different timing of the emergence of WNV L1 and L2 strains from Africa33, which in turn may be due to an apparently lower tendency of WNV L2 strains to spread. For this reason, WNV L2 may appear to be more conserved than WNV L1 Clade 1A strains.
Looking at the two major Eastern and Western phylogeography and genomic epidemiology of WNV L1 and L2 strains between Africa and Europe, they interestingly overlap with two most important Afro-Palaearctic bird migration flyways34, suggesting a possible role of migratory bird species as carriers of WNV across different geographical areas. This scenario is supported by several studies showing WNV to be carried around by migratory birds2,6,10,35, which move across the globe (https://migrationatlas.org/) spreading the virus from their original niches to new areas during this natural process8. In fact, during their annual cycle, birds cross the borders of numerous countries with multiple stopovers while heading to their breeding and non-breeding grounds36. At stopover sites, the virus can spread and become established depending on the composition of the mosquito and resident bird communities6,37. Each bird species appears to be characterised by a variability in migration patterns, creating a complex network of connectivity between Africa and Europe (https://migrationatlas.org/), which may underlie the enormous and variable spread of WNV L1 and L2 strains worldwide. However, it is still difficult to understand the reason why WNV L1 spreads more efficiently than L2, given that both lineages have the same chance of infecting bird species. One possible explanation could be that WNV L1 and L2 infection may have different outcomes in these species5,38. For instance, WNV L2 infection could result in a severe and fatal disease or in a very mild infection with brief and low levels of viraemia39,40,41,42. In either case, the host would decrease its capability to transmit and spread the infection.
When investigating the diverse pattern of circulation of the two viral lineages, it is important to consider the involvement of vectors in viral dispersal. In fact, vectors play a crucial role in transmitting viruses during their blood meal43. Most human outbreaks worldwide have been caused by L1 strains despite the strong presence of L2 in Europe and some African countries10,15,31,32,43, and the existence of other local strains, such as L8 and KOUTV in Africa44,45, or the Rabensburg virus and L4 in Europe46,47. While WNV L1 and L2 seem to have a minor impact in Africa10, the introduction of these African strains in Europe have caused a significant number of severe WNV cases in humans and other animals31,32,43, showing that local strains could have the ability to adapt to new environments and ecological niches and therefore be transmitted and become established. Based on the geographic area, vector species, such as mosquitoes, vary. Depending on factors such as genetic variation, environmental conditions, and evolutionary pressure, different geographic populations are characterised by a diverse behaviour and susceptibility to arbovirus infection48. In Africa, Gamou Fall et al. compared the L1 and L2 vector competence of African Cx. neavei and Cx. quinquefasciatus mosquitoes, often found naturally infected by WNV in Senegal, showing better transmission of L1 compared to L245. In Europe, competent studies performed on Cx. pipiens mosquitoes, the major WNV competent vectors in the continent, showed these mosquitoes to be able transmitters of both L1 and L2 strains49. It clearly appears that the differential circulation of these two strains in Europe and Africa is correlated to multiple factors, as the ability of a mosquito to transmit the virus, the mosquito population density, the host feeding preferences, the availability of susceptible amplifying hosts, the virus divergence coming from the interaction between the virus and the avian host’s immune responses; the diverse pathogenicity of circulating strains; and the association with human and animal populations48.
Mapping lineage dispersal can be an important tool to help identify potential sources of infections, track the direction and speed of spread, and reveal areas of high transmission or disease hotspots, offering valuable information for epidemiological investigations and shedding light on the ecological and environmental processes. Our work constitutes a step in this direction. It gives an integration of extensive and diverse data obtained both from Italy and West Africa (Senegal), and reinforces prior studies and findings highlighting the genetic relationships and circulation dynamics of the two strains between European and African countries. In particular, we collected strains that are genetically distinct from the ones publicly available (e.g. ON813216|Senegal|2012/08/31* and ON813215|Senegal|2012/08/31* for WNV L1 and OQ204315|Italy|2022/11/02* for WNV L2), which allow us to improve the final phylogenetic and phylogeographic inference. By analysing the evolutionary relationships of the virus through phylogenetics and incorporating spatial information through phylogeography, we gain valuable insights into how the two viral lineages have independently evolved and dispersed across space and time. This knowledge has practical implications for surveillance and preparedness efforts concerning future outbreaks of WNV and other viruses, enhancing our ability to monitor and respond to WNV outbreaks effectively, and ultimately contributing to public health and disease control strategies.
While in this study we used a comprehensive collection of WNV L1 and L2 genomes, the limited surveillance systems of some European and African countries lead to gaps in WNV genomic surveillance data, with a limited number of newly generated genomes and sequences available mostly for certain areas and totally absent for other areas. There is still value in conducting analyses based on even larger datasets that cover regions that have been underrepresented in our current study. Expanding the dataset to include samples from these regions will enhance the robustness and reliability of future analyses, providing a more comprehensive understanding of WNV L1 and L2 dispersal dynamics and helping identify potential routes of transmission across diverse geographic regions. Moreover, the absence of precise metadata associated with viral genomes, specifically the collection date and sample location, often unavailable or unreported, constitute a further limit. Having accurate geographic coordinates and collection dates for isolates collected over a significant period of time is essential for precisely calibrating molecular clock and phylogeographic models. This accuracy allows for dependable estimation of when and where epidemic events occurred. To address this challenge, we transformed descriptive sampling locations (like districts, provinces, or regions) into their corresponding geographic coordinates. Additionally, we addressed cases where collection dates were missing by attempting to retrieve them from the relevant research papers, if available. In situations where the collection date couldn’t be identified, we assigned an estimated date. If at least the year was known, we selected the average date calculated from samples with complete information. However, it is vital to systematically ensure the availability of precise location and time metadata to allow a comprehensive phylogeographic and molecular clock analyses of viral epidemics.
Overall, the enhancement of an integrated and homogeneous surveillance plan in Europe and Africa would yield valuable benefits by allowing the generation of new consistent datasets including newly generated genomes and sequence metadata from previously unreported countries and regions. To unravel the L1 and L2 strain dynamics, it would be essential to combine these new datasets with studies on vector competence of diverse WNV strains, alongside with investigations into the abundance and host feeding preferences of various mosquito species. Finally, incorporating GPS data from migratory birds and studies on different bird species susceptibility would further contribute to the understanding on how the virus spread and which reservoirs play a significant role in its transmission across different regions, helping to mitigate the impact and predict future outbreaks, not only of this pathogen, but also of new emerging viruses.
Source link : https://www.nature.com/articles/s41467-023-42185-7
Author :
Publish date : 2023-10-13 07:00:00
Copyright for syndicated content belongs to the linked Source.